Gapped Alignment Phase
Once a collection of ungapped alignments has been found, the next step is to turn a subset of those alignments into gapped alignments, then to keep the highest-scoring subset of those. Blast did not have gapped alignments in mind when it was originally written, and we've expended a significant amount of work to make the current engine handle gapped alignments in a clear and flexible way.
Gapped alignments are handled in two distinct phases. The first is a score-only phase, which only produces the endpoints and the score of each gapped alignment. The more expensive traceback phase actually recomputes all of the work performed in the score-only phase, with different parameters that cause the code to expend more work to produce a better alignment. This time, however, the alignment code generates enough bookkeeping information to allow later stages to reconstruct each alignment in full. blast_gapalign.c contains all of the low-level alignment routines and the top-level routines for the score-only phase, while blast_traceback.c contains the top-level routines for the traceback gapped alignment phase.
The end result of the gapped phase, both score-only and traceback, is a BlastHSPList (blast_hits.h) containing among other things a collection of BlastHSP structures. At the end of the score-only and traceback phases, the HSPs within the BlastHSPList are sorted in order of decreasing alignment score (i.e. in order of decreasing BlastHSP::score). A BlastHSPList contains all of the alignments between one query sequence and one database sequence.
The input to the score-only phase is a BlastInitHitList (blast_extend.h) containing a list of ungapped alignments. These may just be hits from the lookup table that are forwarded to the gapped phase. It's expected (though not necessary) that each BlastInitHSP structure within the BlastInitHitList contain a filled-in ungapped_data field, which gives the extent and score of the corresponding ungapped alignment. Any gapped alignments performed must begin from one of these ungapped alignments. The ungapped alignments in the list are sorted in order of decreasing score.
The main routine controlling gapped alignments for all blast searches except phiblast is BLAST_GetGappedScore (blast_gapalign.c), while the routine for computing gapped alignments with traceback is Blast_TracebackFromHSPList (blast_traceback.c). Because gapped alignments are expensive, these routines go to a lot of trouble to avoid computing them unless absolutely necessary. The procedure is as follows:
- The main alignment procedure:
for (ungapped alignment i) {
-
locate the one query context, in the concatenated set of queries, that contains alignment i; change the query offsets of alignment i to be local to this context. If any previously saved gapped alignments completely envelop alignment i, delete alignment i; the assumption is that the previous alignment was optimal, and alignment i would not add new information if extended
-
call
BlastGetStartForGappedAlignment, which finds the highest-scoring group ofHSP_MAX_WINDOW(=11) contiguous letters in alignment i. The middle of this region is the start point for the gapped alignment. Note that this does not happen for nucleotide searches; in that case, the start point for the ungapped alignment is assumed to be a group of at least four exact matches between query and subject sequences (the original seed hit). There is no 'better' or 'worse' start point within the seed hit; for greedy nucleotide alignment the seed is used directly, while for non-greedy gapped alignment, the start point is the first byte within the match region that contains only exact matches -
(traceback only) call
AdjustSubjectRange. If the subject sequence is smaller thanMAX_SUBJECT_OFFSET(=90000) letters, this does nothing. For larger subject sequences, this routine artificially limits the range of the subject sequence that will be seen by the gapped alignment code. The limitation is not that strict: extension to the right is limited to the distance to the end of the query sequence from the query start point, plusMAX_TOTAL_GAPS(=3000) letters. Extension to the left is similarly limited. This basically assumes that no gapped alignment will have more than2\*MAX_TOTAL_GAPSgaps in the query sequence. Of course if the query is also large the range becomes the whole subject sequence instead.
It's not clear that this measure is needed anymore; we do not actually retrieve partial subject sequences from blast databases, and about the only advantage to having it is that when the query is small it places an upper bound on the size of the auxiliary structures used in the gapped alignment. Because these structures now grow dynamically, a limit probably isn't necessary, but then again it's not performance critical and it's not much of a limit.
- convert alignment i into a gapped alignment. If the resulting score exceeds a cutoff, save the gapped alignment in a
BlastHSPstructure
}
-
Once all of the HSPs are computed, delete any HSPs what share a starting or ending (query,subject) pair with a higher-scoring HSP. The routine that does this is
Blast_HSPListPurgeHSPsWithCommonEndpoints(blast_hits.c) -
Sort the surviving HSPs by score before they are forwarded to the next phase
For score-only alignments, steps 2 and 3 happen in blast_engine.c
The second step in the loop, testing to see whether alignments are contained within other alignments, is performed using a binary-tree-like data structure called an interval tree, described in Appendix B. For protein searches, ungapped HSPs are discarded if the query range and subject range are both enveloped by the query and subject ranges of a previously found gapped alignment. For contiguous megablast, the containment tests are a little more subtle: ungapped alignments may lie inside the 'box' formed by a higher-scoring previous gapped alignment, but must lie more than MB_DIAG_CLOSE (=6) diagonals away from that gapped alignment (see the MB_HSP_CLOSE macro of blast_gapalign_priv.h).
There are two reasons for this difference; first, gapped alignments found by megablast can be very large, and the greedy nature of most megablast gapped alignments means that smaller gapped alignments can often cluster around interesting regions of the main, 'big' alignment. Second, since most gapped alignments have few actual gaps, older versions of this code implemented containment tests by sorting the list of gapped HSPs in order of increasing diagonal and limiting the search to HSPs up to MB_DIAG_CLOSE diagonals away. This was much faster than the alternative, testing every ungapped HSP against all previously computed gapped alignments. Using an interval tree for this application allows the two kinds of containment tests to be unified, and for very large lists of alignments is much faster still.
A note about GetStartForGappedAlignment: when this routine computes the start point for the gapped alignment, that start point is saved in the HSP 'gapped_start' field. The assumption is that the traceback version of the alignment is also justified in starting at this point. For protein blast this is safe to assume, but for nucleotide blast it's only an approximation. The problem is that during the score-only phase, the subject sequence is compressed for nucleotide blast, so we don't know where ambiguities may be in the alignment. These ambiguities may adversely affect the score at the point where the gapped alignment is chosen to start. Thus, the traceback phase first calls BLAST_CheckStartForGappedAlignment, which verifies that when ambiguities are present in the subject sequence, the previously saved start point for the alignment still has positive score in a region of size HSP_MAX_WINDOW centered at the start point.
If this is not the case, then we're stuck; only the boundaries of the alignment are known, so there's really no a priori good way to get a good alignment start point. Currently the code calls BlastGetStartForGappedAlignment from the left end of the gapped alignment. This isn't a bad idea, since a good local alignment will not have many gaps and will start and end with a long run of matches. A long run of matches will at least mean that the traceback alignment will start in a locally good region. It's unlikely that the region will have the optimal score, though, since gaps later in the alignment will move it to a different diagonal that could be better scoring. There may be better ideas to solve this problem, but the current procedure works well enough and blast has never really been obsessed with always finding the best possible alignments.
The low-level routines also fill in an input boolean called fence_hit. This represents a hook for the fetching of partial database sequences. The C++ toolkit contains an advanced library ('SeqDB') for accessing sequences from blast databases, and in certain applications (like binaries that generate traceback from previously computed blast results) the extents of all the alignments going into the traceback phase of blast are known. In this case, memory is allocated for database sequences but only the portions that will be used in the traceback phase are actually initialized. The problem is that traceback alignments can become larger than when they started, and there's no way to know how large they will get.
We work around this problem by assuming that all alignments will become X bases larger at most, then putting 'fence' bytes at the boundaries where initialized letters end inside the database sequence. If the traceback gapped alignment routines encounter this fence byte, the boolean is set to true and the entire traceback process fails for this DB sequence. Calling code then retrieves the entire database sequence and reruns the traceback. When the database contains very large sequences, this makes the traceback generation process dramatically faster (5-10x), and since traceback ordinarily consumes a very small fraction of the total runtime, putting a test for the fence byte in the innermost loop does not noticeably reduce throughput.
The following covers the low-level dynamic programming routines, located in blast_gapalign.c.
Gapped alignments may be computed from protein or nucleotide ungapped HSPs. For translated protein searches, routines are also available for computing out-of-frame gapped alignments. Gapped alignments for all protein searches call s_BlastProtGappedAlignment, and non-greedy nucleotide gapped alignments begin with s_BlastDynProgNtGappedAlignment. Both of these routines set up the sequence offsets that describe where the gapped alignment should start, and also handle collection of the ending offsets and scores. Each gapped alignment is composed of a left extension and a right extension; the alignment score is the sum of the scores for these two extensions. Obviously, if the start point is at the boundary of one sequence or the other then a gapped extension cannot begin in that direction.
All gapped extensions, whether to left or right, compute the offset into both sequences where the extension ends, and the score of that extension. They can optionally also produce the complete traceback for the alignment. Alignment extents and scores fill in the fields of a BlastGapAlignStruct (blast_gapalign.h), from which calling code disperses the information to other structures.
There are five main low-level gapped routines:
-
ALIGN_EXperforms gapped alignment of two protein sequences, or two nucleotide sequences in ncbina format. Computes the alignment traceback -
Blast_SemiGappedAlignperforms a gapped alignment of two protein sequences -
s_OutOfFrameAlignWithTracebackperforms a gapped alignment of a protein sequence with an out-of-frame protein sequence, and computes the alignment traceback -
s_OutOfFrameGappedAlignperforms a gapped alignment of a protein sequence with an out-of-frame protein sequence -
s_BlastAlignPackedNuclperforms a gapped alignment of a nucleotide sequence in blastna format with a packed nucleotide sequence in ncbi2na format.
All of these routines perform extensions to either the right or to the left. blastn and discontiguous megablast searches use s_BlastAlignPackedNucl for the score-only gapped alignment and ALIGN_EX for the traceback alignment; by the time traceback happens, all sequences are in one-letter-per-byte form, and this is all that ALIGN_EX really needs (along with a suitable score matrix). The out-of-frame routines always assume that the second input sequence is the out-of-frame one. This means that for blastx searches calling code has to swap the inputs. There is no out-of-frame routine that allows frame shifts in both sequences; this would be much more computationally intensive, and it's not clear that the extra alignments found by 'OOF tblastx' would actually be useful. Finally, the first two routines are not static because the composition-based statistics code in blast_kappa.c uses them directly.
The low-level details of how these routines work is beyond the scope of this document. Some highlights of why they look the way they do can help understand them, though.
The in-frame routines are variants of ordinary Needleman-Wunsch dynamic programming. s_BlastAlignPackedNucl is essentially identical to Blast_SemiGappedAlign, except that the first input sequence is unpacked letter by letter as the extension takes place. There are several variations in these routines that are unusual:
-
All the gapped routines incorporate an X-drop criterion. The dynamic programming lattice is filled in row by row, and if the best possible score in a given lattice cell is less than some number X below the best score seen thus far, the best score at the cell is set to -infinity (guaranteeing that the final alignment will not pass through it).
-
This means that a given row can only explore a few cells beyond the extent of the previous row; it also means that if the first cells in the row fail the X-drop test, the next row will begin some distance past the current row. Hence the portion of the search space that the gapped extension (in one direction) examines usually looks like the following (this corresponds to a gapped extension to the right of the start point):
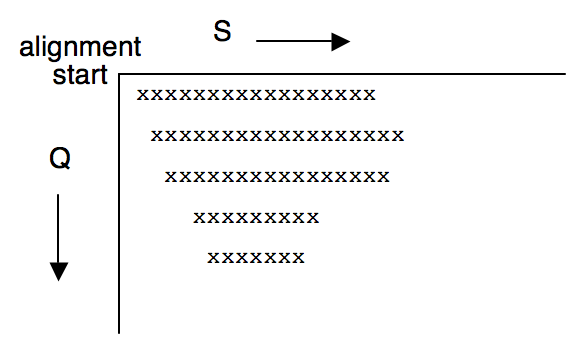
There is a very unintuitive side effect to the X-drop test when implemented this way. If the region that does not fail the X-drop test instead looks like:
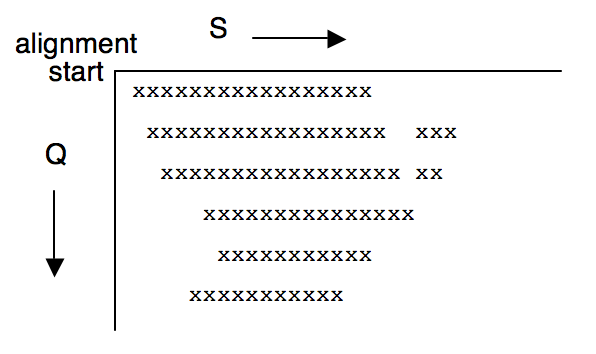
i.e. has 'horns' or concave regions, then the horn in the second and third row gets explored (because the number of cells to examine is determined before the row starts) but the horn in the last two rows is ignored because the starting cell of a row always increases monotonically. In general, this means that a gapped alignment of sequence A and B may not turn out to be equivalent (even in score) to a gapped alignment of sequence B and A, despite having the same start point.
- The factor that adds the most complexity to the code boils down to the fact that the input sequences can be very large. Most Needleman-Wunsch implementations assume that at least a linear array of per-letter structures is acceptable, but these routines cannot. If one sequence has 10 million nucleotides, then a traceback gapped alignment would need hundreds of megabytes for the auxiliary structures needed during the dynamic programming, even if the actual alignment was only 100 letters long and needed a few kilobytes for the actual traceback. Hence all of the auxiliary structures are allocated dynamically, and only increase in size if the alignment demands it. The array of auxiliary structures is reused from subject sequence to subject sequence. For traceback alignments, the space allocated for the auxiliary data needed to reconstruct the traceback uses a separate memory manager, since many small allocations and no deallocations are expected. The same considerations apply to protein sequences, even if most protein sequences are small: a translated search can turn a giant nucleotide sequence into several giant protein sequences.
For contiguous megablast, alignments are expected to be very large but to contain few gaps. For that situation an ordinary dynamic programming gapped alignment will do too much work, since it makes no assumptions about how gap-filled the optimal alignment is. For this type of problem, blast uses specialized routines located in greedy_align.c that attempt to skip over large portions of the ordinary dynamic programming search space in an attempt to build an optimal gapped alignment greedily. These routines are called from BLAST_GreedyGappedAlignment (blast_gapalign.c) and only care about matches, mismatches and gaps; X-dropoff criteria are still used, and the methods are parametrized to perform a configurable amount of work looking for an optimal alignment.
A low-level understanding of how the greedy routines work would require a careful reading of the paper that originally described them, as well as of the code in the current implementation. Briefly, ordinary dynamic programming uses a table that corresponds to aligning a portion of both sequences; each cell of the table gives the score of an optimal path ending at that cell. Greedy methods instead measure an alignment in terms of a 'distance', i.e. the combined number of gaps and of mismatches that occur on that path. Basically the routines assume that an alignment score depends only on its length.
The table for a greedy algorithm assigns distances to each row and a diagonal to each column. Each cell in this table contains not a score, but the maximum offset into one of the sequences that lies on the diagonal and achieves the distance described by the cell. The larger the offset, the more matches occur in the alignment and the higher its score (i.e. for fixed distance and diagonal, how many matches can be achieved?). The greedy algorithm examines each distance in turn, and stops when any larger distances would fail an X-dropoff test, or at any rate when a maximum distance has been processed. For each distance, it then iterates through all of the diagonals that were valid in the previous distance, looking for the diagonal that can achieve the current distance with the largest number of matches. This is a 3-term recurrence relation that looks a lot like ordinary dynamic programming. The number of diagonals examined can increase or decrease when examining the next distance. For each distance, the score of the best path is saved separately; a traceback procedure can use the table of sequence offsets to create a list of edit operations that recover the optimal alignment.
The number of distances examined when aligning sequences A and B is either GREEDY_MAX_COST (=1000) or ((length of B) / 2 + 1), whichever is smaller. In the implementation the maximum number of diagonals to examine can only increase by at most 2 for each distance, so this also caps the maximum number of diagonals. Roughly speaking, no matter how large the input sequences are, the alignment produced will never venture more than a certain number of mismatches/gaps away from 'all matches'. Unlike a 'banded aligner', which has the same restriction, the greedy aligner does not need to examine every combination of sequence offsets that both lies in the band and passes the X-drop test.
The paper describing the algorithm claimed order-of-magnitude speedups as a result; my personal opinion is that the big advantage of the greedy routines is not performance (for ordinary alignments they do not appear to be materially faster than the ordinary gapped routines we now use) but memory efficiency. The central idea of the greedy alignment routines is that they implement a runlength encoding of the ordinary dynamic programming lattice; if your gapped alignment is a million letters long but only has three actual gaps, the greedy routines only have to store perhaps a few hundred states.
There are two species of greedy alignment supported, one with affine and the other with non-affine (linear) gap costs. BLAST_GreedyAlign implements the linear version; this requires gaps to have a constant per-letter penalty of exactly (mismatch_score - match_score / 2). This restriction lets the aligner get away with less than 1/3 the memory consumption and arithmetic of the affine aligner, but the alignments it produces are most suited to cases where the only reason to have a gap is because of sequencing errors, i.e. the alignments are not expected to be biologically plausible. The affine greedy code is in BLAST_AffineGreedyAlign, and here the match, mismatch, gap open and gap extension penalties are arbitrary. Zero gap penalties are a signal to the affine routine that the non-affine routine is to be used; the non-affine routine is never called directly. Note that the standard paper that describes greedy alignment only goes into detail about the case of linear gap penalties.
Unlike the ordinary dynamic programming alignment code, there is no separate version of the affine and non-affine greedy routines that calculates traceback. The traceback can optionally be computed right after the alignment bounds are determined. In the current engine, traceback is performed in a separate call to the greedy aligners, with a more generous X-drop criterion (and ambiguities in the subject sequence represented faithfully). This has the nice advantage of stitching together many small alignments into a single big one when the underlying neighborhoods of the query and subject sequence are very similar to each other. It also allows gapping across stretches of ambiguities that would have split apart a score-only version of the alignment.
Occaisionally, when ambiguities are restored they invalidate the start point that is computed during score-only greedy alignment. This usually means the traceback will start from a suboptimal point and generate spurious gaps to rejoin the otherwise optimal alignment. To make this less likely, the low-level greedy routines remember the longest run of exact matches encountered during the gapped extension, and return that information in an SGreedySeed (greedy_align.h) structure. When score-only alignment completes, the alignment start point is chosen as the middle of this region. Recovering the longest run information is easy because the greedy recurrence relations need the same information anyway.
For nucleotide blast, the time needed for gapped alignment is usually not that significant. However, for protein blast experiments show that gapped alignment takes a fairly consistent 25-30% of the total runtime, and the vast majority of that time goes into the score-only gapped alignment phase. Cameron/Williams/Cannane from RMIT developed a clever method for reducing the score-only gapped alignment time, and a modification of this scheme is implemented in the NCBI blast code.
Ordinary gapped alignment computes a 3-term recurrence relation for every cell of the dynamic programming lattice. The central idea with the modified scheme is that if gaps are only allowed to start at specific offsets in the sequences being aligned, then most cells in the lattice can be computed using a 1-term or (occasionally) 2-term recurrence. The drawback is that because gaps may not be allowed to start at the point needed to achieve an optimal alignment score, the score from this 'semi-gapped' alignment may be worse than the true optimal score one is entitled to. CWC's idea was to first perform a semi-gapped procedure on each ungapped alignment that needs extending. If the score achieved exceeds the gapped cutoff score, then the alignment bounds and the score are kept. If the score cannot exceed a reduced cutoff score (set to 68% of the gapped cutoff), the alignment is thrown away. If the alignment achieves a score between these two extremes, it is recomputed using the full optimal score-only alignment.
A very nice advantage to this scheme is that as the cutoff for the search increases, more and more gapped alignments are computed using the semi-gapped algotithm, i.e. the optimal score-only routine is needed less and less often and performance increases. Without semi-gapped alignment, performance increases a little but that's only because fewer ungapped alignments make the more stringent ungapped cutoff to begin with. Semi-gapped alignment gives a boost over and above this.
Semi-gapped alignment is performed by s_RestrictedGappedAlign. This routine is approximately five times faster than Blast_SemiGappedAlign, since its inner loops are much simpler most of the time. Implementing the full CWC scheme speeds up blastp by about 5% with an e-value cutoff of 10.0; as the cutoff becomes more stringent, the speedup climbs to about 15% asymptotically. With the semi-gapped routine being five times faster, the largest speedup we can expect is about 22%, so the performance behaves about as expected.
Unfortunately, implementing CWC's scheme as described causes huge differences in the output. If all the hits expected to be found are high-scoring, the differences are minor; but if some low-scoring hits are expected, many of them are missed. More importantly, some hits (including high-scoring hits) do appear in the output but end up scoring lower and looking worse, with big gapped regions that did not originally appear.
Each score-only gapped alignment has a start point, and the traceback phase reuses that start point. Starting the traceback alignment from a suboptimal region will produce a lower-scoring final alignment, and the differences come about because some alignments are allowed to graduate to the traceback phase that previously did not do so. These alignments carry their suboptimal start point with them; the reason is that CWC's scheme produces a list of HSPs that are computed by a mixture of approximate and exact gapped alignment. If the approximate alignment is good enough, it is not recomputed. Other alignments may not be good enough either if computed approximately, but sometimes an approximate alignment gets recomputed optimally. When this happens, a mediocre alignment computed optimally will sometimes trump a better alignment that was computed approximately. This causes the mediocre alignment to erroneously make it to the traceback phase.
This means that we cannot mix together approximate and exact gapped alignments. All alignments for a given HSP list should be computed approximately, and if the score from one of those alignments is ambiguous then all of the previously computed alignments should be thrown away and recomputed optimally. This technique removes 90% of the output differences, but the total number of gapped alignments increases, and wipes out the runtime savings of using semi-gapped alignment at the default e-value cutoff! We can recover the performance with one more trick: semi-gapped alignment is not attempted at all if the highest-scoring ungapped alignment exceeds the gapped cutoff. Experiments show that when this happens, there is usually another ungapped alignment in the list whose score will be ambiguous if we try to extend it using the semi-gapped code, in turn causing all the previously computed approximate alignments to be thrown away. Basically this change uses semi-gapped alignment only when all the ungapped alignments are mediocre, and this avoids computing something like 80% of the approximate alignments that would be thrown away later.
These changes mean that the output differences are minor, and we still get a 5-10% speedup of blastp. That doesn't sound like much, given all of the complexity of the new alignment method, but after years of development there really isn't all that much room for performance improvement in blastp, and we'll take any improvement we can get.
The functions in blast_sw.[ch] replace all of the alignment algorithms used by the engine with a custom implementation of the Smith-Waterman sequence alignment algorithm. All of the non-sequence-alignment portions of the engine (sequence handling, translation, statistics, etc.) still run normally, but when the engine is configured to perform Smith-Waterman alignment:
-
No ungapped wordfinder runs at all
-
The score-only gapped routine produces at most one HSP per query context. The HSP contains the relevant context, the entire range of the query and subject sequence, and the highest local gapped alignment score found by the score-only Smith-Waterman code. Every context of every query sequence is individually aligned to each subject sequence
-
The Smith-Waterman traceback routine takes each HSP from the score-only phase and converts it to a list of all the local alignments between query and subject sequence whose score exceed the gapped score cutoff, along with the traceback for each alignment. This is the only example in the blast engine where one input alignment generates multiple different output alignments. The Smith-Waterman code has to work this way because the score-only routine examines every query-subject offset pair but only produces one alignment score. The traceback routine does much more bookkeeping, using a clever variation on the basic algorithm to recover all the local alignments one at a time.
Nobody realistically expects the Smith-Waterman code to be used at the NCBI in a production environment. It was added to the blast source for several reasons:
-
The alignment algorithm is rigorous, and does not depend on the heuristics that make blast run fast. It thus can provide 'gold standard' alignment results to which ordinary blast can be compared. Users can also take advantage of the rigorous results when the input problems are known to be very small, and alignment accuracy is vital
-
Smith-Waterman is very easy to implement in dedicated hardware, and adding an implementation to the blast engine should make it easy for hardware companies to insert their own super-optimized Smith-Waterman hardware into an otherwise unmodified code base.
-
the other features in the BLAST engine, i.e. sophisticated statistical models, translated searches, standardized output formatting, etc. are hard to find in other Smith-Waterman programs
Users should be aware that the vast majority of the time the ordinary BLAST heuristic is more than sufficient for your local alignment needs. The Smith-Waterman implementation runs 50-100 times slower than ordinary BLAST, and the alignments found are identical unless the alignment scores are very low. This is hardly surprising; BLAST has been optimized and tuned for almost 20 years now, and has always been intended as a complete replacement for Smith-Waterman. The code is present more for test purposes than anything else.